Sign up for our newsletter
Join our scientific community to stay up to date with Element news, insights, and product updates.
This site is protected by reCAPTCHA and the Google Privacy Policy and Terms of Service apply.
How to evaluate next-generation sequencing platforms for minimal residual disease research and why error rate, depth, and cost-per-sample determine the limit of detection you can actually reach.
Key takeaways
For minimal residual disease (MRD) detection, sequencing accuracy is an important platform attribute: a lower background error rate is one factor that lowers the variant allele frequency (VAF) you can confidently call. Independent benchmarking from the SMaHT Network found that the Element AVITI™ System had the lowest substitution error rate of any short-read platform in difficult (“non-easy”) genomic regions (0.04%) and the lowest indel error rate in homopolymers (0.14%), enabling accurate somatic SNV discovery down to ~0.3% VAF. Combined with a low cost-per-sample, this lets MRD researchers sequence deeper within a fixed budget, the other lever that sets the detection floor.
What is MRD detection?
Minimal residual disease refers to the small number of cancer cells that can remain in the body after treatment which are often below the threshold of imaging or standard pathology. In MRD research, next-generation sequencing (NGS) is used to detect circulating tumor DNA (ctDNA) or tumor-derived variants at extremely low abundance, frequently far below 1% variant allele frequency (VAF). The goal is to identify residual or recurrent disease earlier than conventional methods, understand treatment response, or study relapse dynamics.
Because the tumor signal is so rare, MRD is one of the most analytically demanding applications in sequencing. An assay that performs well for routine genotyping can fail entirely at MRD-relevant VAFs, where a handful of true variant reads must be separated from sequencing noise. That makes platform selection a sensitivity consideration, even ahead of workflow considerations.
Why is sensitivity the defining requirement for MRD assays?
Sensitivity in MRD research is usually expressed as the limit of detection (LoD), the lowest VAF (or the lowest tumor fraction, often quoted in parts per million), at which the assay reliably distinguishes a true variant from background. Two factors set that floor:
-
Sequencing accuracy: the platform’s background error rate, which determines how much noise sits underneath a true low-VAF call.
-
Depth and molecular recovery: the sequencing depth and molecular coverage achievable per sample, which determines how many unique DNA molecules you actually observe at each position.
These interact directly. To detect a variant at 0.1% VAF, you need enough unique molecules covering that position to see multiple independent variant-supporting reads and a low enough error rate that those reads are not obscured by sequencing artifacts. A platform with high raw accuracy reaches a given LoD at lower depth, which in turn lowers cost per sample. This is why accuracy and economics are linked rather than separate considerations.
Definition: variant allele frequency (VAF)
VAF is the fraction of sequencing reads at a locus that carry the variant allele. A 0.5% VAF means roughly 5 in 1,000 molecules carry the mutation. MRD assays commonly target detection in the 0.01%–1% VAF range, depending on whether the design is tumor-informed or tumor-naive.
What sensitivity do MRD assays actually require?
Requirements vary by assay architecture. Two broad strategies dominate MRD research:
|
Approach |
How it works |
Typical sensitivity target |
|
Tumor-informed (bespoke) |
A personalized panel is designed from the patient’s tumor mutations, then tracked in plasma over time. Many targets are monitored at once. |
~0.001%–0.01% VAF (1–10 ppm) with deep sequencing up to 100,000x coverage and error suppression |
|
Tumor-naive (fixed panel) |
A fixed gene panel is applied without prior tumor knowledge. Useful when tissue is unavailable. |
~0.1%–1% VAF, depending on panel size and depth |
|
Genome-wide/WGS-based |
Low- to moderate-depth whole-genome signal is aggregated across thousands of sites to boost effective sensitivity. |
Relies on aggregate accuracy across the genome, including difficult regions |
Whichever approach a research program uses, the underlying platform must deliver high per-base accuracy and the ability to recover and sequence many unique molecules. Tumor-informed designs lean hardest on raw accuracy because they integrate signal across many low-VAF sites; an elevated error rate at any of those sites inflates false positives.
What determines an NGS platform’s sensitivity for MRD?
When evaluating platforms for MRD research, focus on the factors that move the limit of detection:
-
Raw substitution and indel error rate: Lower background error means fewer artifactual reads masquerading as true low-VAF variants. This is the dominant determinant of LoD before any error-correction is applied.
-
Accuracy in difficult regions: Many MRD panels target homopolymer-adjacent or repetitive regions. Platforms differ enormously here — with long-read chemistries in particular showing high indel error in homopolymers.
-
Compatibility with UMI/consensus methods: Unique molecular identifiers and duplex/consensus strategies suppress errors, but they consume reads. A platform with lower intrinsic error needs less aggressive (and less wasteful) error correction.
-
Depth and coverage uniformity at workable cost: Reaching deep, uniform coverage affordably lets you observe more unique molecules per sample, which directly lowers the achievable VAF.
-
Reproducibility across runs: MRD studies run many samples longitudinally. Run-to-run and replicate consistency is essential for tracking small changes in tumor fraction over time.
How do sequencing platforms compare on accuracy?
Accuracy claims are best assessed with independent, controlled benchmarking. A study from the NIH Somatic Mosaicism across Human Tissues (SMaHT) Network is particularly relevant to MRD because it used naturally occurring, orthogonally validated low-VAF mutations, from 0.1% to 25% VAF, to estimate platform-specific error rates across four major technologies (Jang et al., 2025, preprint).
The headline result for MRD researchers: error rate varies by more than an order of magnitude between platforms, and the gap widens in exactly the difficult regions where MRD panels often operate. Substitution error, the noise floor under a low-VAF SNV, is shown below.
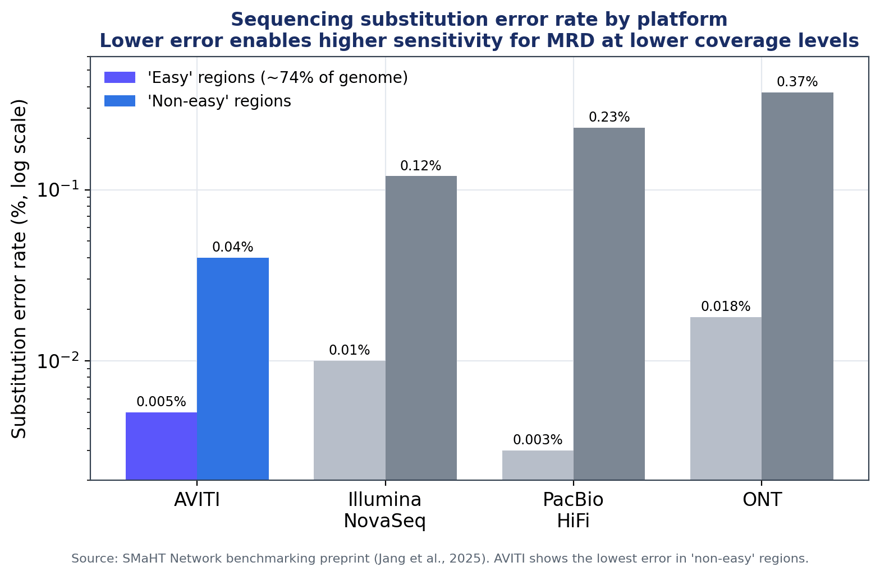
Figure 1. Substitution (SNV) error rate by platform in “easy” vs “non-easy” genomic regions. Lower error supports a lower limit of detection. Data: SMaHT Network benchmarking (Jang et al., 2025).
In the easiest ~74% of the genome, the best short- and long-read platforms are close (AVITI 0.005%, PacBio HiFi 0.003%, NovaSeq 0.010%). But in “non-easy” regions, where many clinically relevant loci sit, AVITI had the lowest error of all platforms tested at 0.04%, roughly 3x lower than NovaSeq (0.12%) and ~6x lower than PacBio (0.23%). For indels, the divergence is higher.
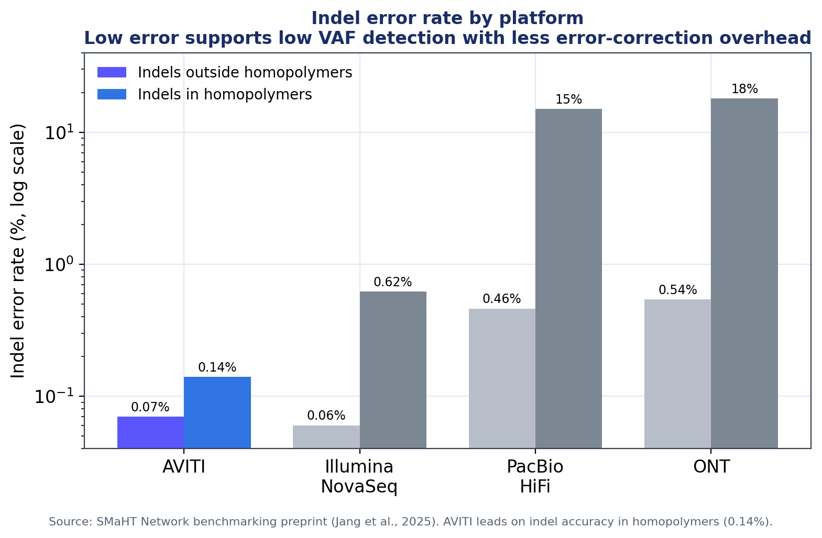
Figure 2. Indel error rate by platform, inside vs outside homopolymers. Long-read platforms show very high homopolymer indel error. Data: SMaHT Network benchmarking (Jang et al., 2025).
AVITI showed the lowest indel error in homopolymers (0.14%) among all platforms—versus 0.62% for NovaSeq and roughly 15–18% for the long-read platforms, where homopolymer indels were effectively indistinguishable from background. For MRD panels that include indel targets or homopolymer-adjacent sites, this difference can be important.
WHY THIS MATTERS FOR MRD
The benchmarking authors reported accurate, specific somatic SNV discovery below 1% and as low as ~0.3% VAF, with non-homopolymer indels reliable down to ~1% VAF. A platform’s error rate is the practical floor on how low you can push a low-VAF MRD call before false positives create too much noise.
Platform comparison summary
|
Metric |
AVITI |
Illumina NovaSeq |
PacBio HiFi |
ONT |
|
SNV error — easy regions |
0.005% |
0.010% |
0.003% |
0.018% |
|
SNV error — non-easy regions |
0.04% |
0.12% |
0.23% |
0.37% |
|
Indel error — outside homopolymers |
0.07% |
0.06% |
0.46% |
0.54% |
|
Indel error — in homopolymers |
0.14% |
0.62% |
~15% |
~18% |
Source: SMaHT Network benchmarking preprint (Jang et al., 2025). Long-read homopolymer indel values are approximate, reflecting that calls were largely indistinguishable from background.
How does accuracy translate into real low-VAF detection?
Benchmark error rates predict potential; applied studies confirm it. In clinical-research validation work and partner studies, high raw accuracy on the AVITI platform has translated into reliable low-VAF detection across real sample types:
- cfDNA / liquid biopsy: In liquid-biopsy research using the MSK-ACCESS panel for cfDNA, clinical samples sequenced on AVITI showed highly consistent variant fractions, including detection of known variants down to 0.5% VAF.
- Concordance with established platforms: AstraZeneca scientists reported matched variant-calling performance between AVITI and NovaSeq 6000, including accurate detection of low-VAF mutations, small-variant sensitivity and precision, and CNV calling, even using a pipeline originally optimized for a different chemistry.
- Targeted capture reproducibility: Using Trinity™ on-flow-cell target capture, researchers at the Garvan Genomics Platform reported >95% SNP detection sensitivity for heterogeneous variants and VAF correlation of R² ≈ 0.996 between capture workflows on cfDNA and FFPE reference and banked patient samples.
The throughline: when the intrinsic error rate is low, the assay reaches MRD-relevant VAFs with low error-correction overhead and results stay consistent across replicates and runs needed for longitudinal MRD monitoring.
What about cost and scale?
Sensitivity has an economic dimension. Because sequencing depth enhances the achievable VAF, the cost per gigabase determines how deep you can sequence within a fixed cost-per-sample budget. A lower cost-per-sample is, in effect, a sensitivity lever for high-volume MRD programs.
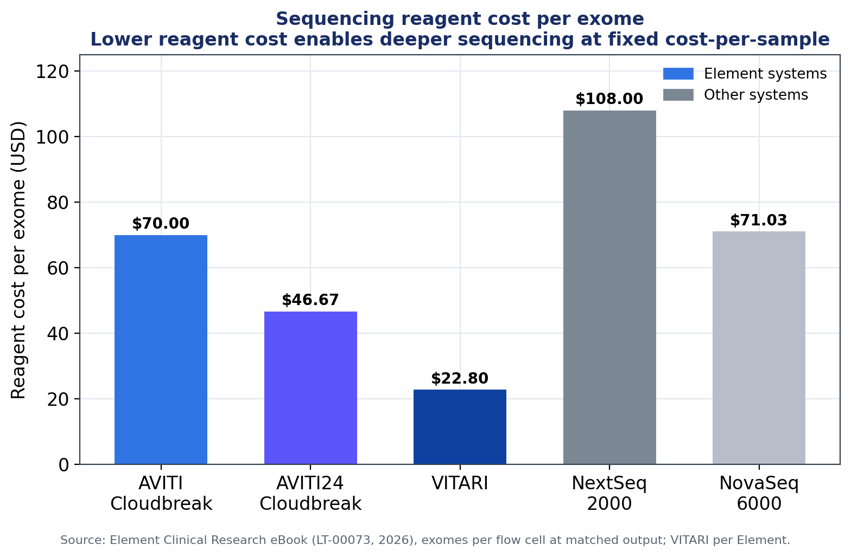
Figure 3. Reagent cost per exome across sequencing systems at matched output. Data: Element Clinical Research eBook (LT-00073, 2026).
On a cost-per-exome basis, AVITI24™ with Cloudbreak Freestyle™ chemistry came in at $46.67 — below NextSeq 2000 ($108.00) and NovaSeq 6000 ($71.03) in Element’s published comparison. Our forthcoming VITARI™ System will further reduce costs for customers operating at higher volumes. Lower reagent cost per sample frees budget for the deeper sequencing that MRD detection demands, and the dual-lane flow cell design supports running multiple panels with different depth requirements in a single run, useful for mixed MRD and CGP workloads. Avidite Base Chemistry™ underpins the accuracy: it was independently shown to deliver Q40-level accuracy (Arslan et al., Nature Biotechnology, 2023).
How to evaluate a platform for your MRD assay: a checklist
When comparing options for an MRD research workflow, work through these questions:
-
Error rate where it counts. Ask for substitution and indel error rates broken out by region type (easy vs difficult, in/out of homopolymers)—not just an average Q-score.
-
Demonstrated LoD on relevant samples. Look for low-VAF detection data on the sample type you care about (cfDNA, FFPE, hematologic), ideally with orthogonal confirmation.
-
Depth economics. Calculate cost per sample at the depth your LoD requires—not list price per run.
-
Error-correction fit. Confirm compatibility with the UMI/duplex/consensus method your assay uses, and how many reads that overhead consumes.
-
Reproducibility evidence. Request replicate and run-to-run VAF correlation, since longitudinal MRD tracking depends on it.
-
Workflow and panel compatibility. Check that your existing capture panels, library prep, and analysis pipelines are supported to avoid revalidation.
Where the AVITI platform fits in MRD research
Element designed the AVITI family of systems around the two requirements that govern MRD sensitivity: accuracy and depth economics. Independent benchmarking places AVITI at the lowest error rate among short-read platforms in difficult regions and homopolymer indels while Cloudbreak Freestyle chemistry delivers Q40 data at a competitive cost per sample. Trinity on-flow-cell target capture adds a streamlined enrichment workflow that preserves accuracy and coverage uniformity, and the platform integrates with established MRD and CGP panels (including SOPHiA MSK-ACCESS and MSK-IMPACT, Twist, IDT xGen, QIAGEN xHyb, and Agilent SureSelect workflows) so labs can adopt it without rebuilding validated pipelines.
For MRD research teams, the practical implication is that high analytical sensitivity and a workable per-sample budget no longer have to be traded off against each other.
Frequently asked questions
What VAF can NGS detect for MRD?
It depends on assay design and depth. Tumor-naive fixed panels typically target ~0.1–1% VAF, while tumor-informed and error-suppressed designs can reach ~0.001–0.01% VAF (1–10 ppm). Independent benchmarking has shown accurate somatic SNV discovery down to ~0.3% VAF and non-homopolymer indels down to ~1% VAF using high-accuracy short-read data.
Which sequencing platform is most accurate for low-VAF detection?
In SMaHT Network benchmarking, the Element AVITI System had the lowest substitution error rate in difficult genomic regions (0.04%) and the lowest indel error in homopolymers (0.14%) of the four platforms tested. PacBio HiFi was marginally lower in “easy” regions for SNVs (0.003% vs 0.005%) but substantially higher elsewhere, especially for indels.
Why does error rate matter more than read length for MRD?
MRD detection is about separating a rare true signal from background noise at a single base. Error rate sets that noise floor directly. Read length helps with mapping and phasing but does not lower the per-base background that limits low-VAF calling and long-read platforms currently carry higher per-base error, particularly for indels.
Do I need UMIs or duplex sequencing for MRD?
For the lowest VAF targets (tumor-informed MRD), molecular error correction such as UMIs or duplex/consensus methods is standard. A platform with low intrinsic error reaches a given LoD with less error-correction overhead, preserving usable reads and lowering cost per informative molecule.
Explore the data and sources
Make your own evaluation with these primary data sources and public datasets:
- Targeted sequencing for clinical research (MRD, CGP, exome) on AVITI
- Dataset: Twist Oncology CGP Panel, Trinity Freestyle™ workflow, 2x150 sequencing
- Dataset: Targeted sequencing with xGen™ Pan-Cancer Hyb Panel (Trinity™ kit)
- Webinar: Rapid, Low-Input CGP Across Tissue and cfDNA Samples at the Garvan Institute
- Webinar: Robust CGP of FFPE Samples Using QIAgen xHYB for Trinity CGP Workflow
- Webinar: Advancing Targeted Sequencing for Clinical Research at AstraZeneca with On-Flow Cell Enrichment
References
1. The Somatic Mosaicism across Human Tissues Network, Alexej Abyzov. Comprehensive benchmarking of somatic mutation detection by the SMaHT Network. bioRxiv 2025.10.09.678885; doi: https://doi.org/10.1101/2025.10.09.678885.
For Research Use Only. Not for use in diagnostic procedures. Benchmarking figures cited from the SMaHT Network preprint reflect a non–peer-reviewed study at time of writing. Third-party product names are trademarks of their respective owners.