Question:
What are some best practices for library quantification before sequencing?
Answer:
Achieving optimal sequencing results depends on accurate library quantification and appropriate loading concentrations. Several factors can affect quantification accuracy and ultimately influence run output, including the chosen quantification method, library size, adapter dimers, overamplification, and variability in sample handling and laboratory technique.
Summary
Best practices for library quantification:
- Quantify libraries before sequencing using the recommended quantification method for your library preparation
- Avoid overamplification
- Check for and remove excess adapter dimers
Considerations
Quantification Methods
Multiple library quantification methods are available, and certain methods can be better suited to specific library preparations. Follow the manufacturer's library preparation recommendations for library QC.
Library Concentration
A general rule for linear libraries is to quantify PCR-free libraries by qPCR and PCR-plus libraries with a Qubit Fluorometer.
If using a Qubit assay, select the appropriate assay based on target molecule and expected sample concentration. Use the Qubit dsDNA HS (High Sensitivity) assay for low-concentration samples (0.2–100 ng/µl). Follow the manufacturers protocol to prepare samples. Important techniques to abide by include:
- Ensuring all materials and reagents are at room temperature before use
- Preparing fresh working solutions in clean plastic tubes
- Vortexing Qubit standards and samples before pipetting
- Letting the samples incubate with the dye protected from light before measuring
For qPCR quantification, preparing multiple small dilutions is preferable to a single large dilution. Use at least two dilution steps where no less than 2 µl are pipetted between dilutions. Ensure each dilution is thoroughly vortexed and spun down before taking the next aliquot to minimize carryover effect and ensure accuracy. If you are unsure of the original sample concentration or the dilution required to be in the middle of the standard curve, you can run multiple dilutions of the same sample (e.g., dilute 1:10 5x to generate 1:10 to 1:10,000 dilutions and quantify 1:100, 1:1000, and 1:10,000).
Reference the Element Adept™ Library Compatibility Workflow User Guide or Element Elevate™ Library Prep User Guide for instructions on assay setup. Additional best practices for qPCR include:
- Mixing master mixes by pipetting, inverting, or flicking (avoid vortex mixing) followed by a gentle spin down
- Keeping master mixes, samples, and qPCR plate on ice until use
- Ensuring plates, seals, and qPCR reagents are appropriate for the instrument, and that the instrument is regularly serviced, calibrated, and tested.
Fragment Size Assessment
Size libraries using an Agilent TapeStation or similar device (e.g. Bioanalyzer, Revvity LabChip, or Fragment Analyzer). Libraries that show broad fragment distributions can make sizing and downstream quantification less reliable. For optimal sizing performance, normalize libraries to the same concentration before analysis. Normalization improves sizing accuracy and helps identify adapter dimers, overamplification, or other library abnormalities. Size Adept libraries before circularization and quantify using qPCR.
Library Overamplification
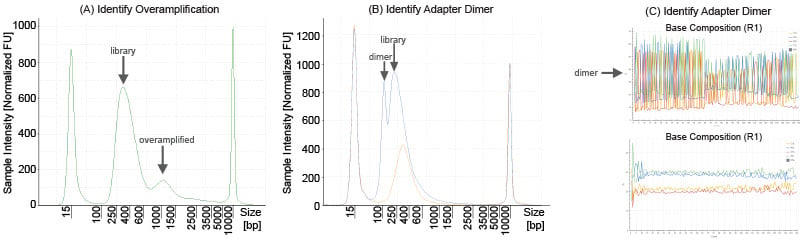
Figure 1. (A) Example QC trace for an overamplified library. (B) Example QC traces for a library with adapter dimer (blue) and without adapter dimer (orange). (C). Example sequencing base composition of a library with (top) and without (bottom) significant adapter dimer.
Library overamplification can distort the library size estimate and result in inaccurate quantification using fluorometric methods. Overamplified libraries create heteroduplex library products as shown in Figure 1A and can only be accurately quantified via qPCR. Avoid using an excess of PCR cycles to prevent library overamplification.
Adapter Dimers
Library quantification can be negatively impacted by the presence of adapter dimers. Adapter dimers can adversely affect sequencing by forming polonies and generating sequencing reads that do not contain a library insert. Before a sequencing run, adapter dimer is generally identified as a peak around 125-170 bp in the library trace, but in some cases can be as low as 100 bp or as high as 200 bp. In Figure 1B, the blue trace illustrates a library with adapter dimer and the orange trace illustrates an identical library where an additional cleanup was performed to remove adapter dimer.
If your TapeStation trace reveals adapter dimers, run an additional bead cleanup before performing final Qubit measurement. The optimal cleanup approach depends on the library type, fragment size distribution, and extent of adapter dimer contamination. Refer to the bead manufacturer's recommendations and your library preparation protocol for guidance.
You can also identify adapter dimer after a sequencing run via “spikiness” in the base composition, especially in read 1 (Figure 1C). However, spikes in base composition are not solely caused by adapter dimer, and knowledge of the expected base composition for the library is helpful to identify whether observed spikes are caused by adapter dimer.