Sign up for our newsletter
Join our scientific community to stay up to date with Element news, insights, and product updates.
This site is protected by reCAPTCHA and the Google Privacy Policy and Terms of Service apply.
In a recent paper published in Nature Communications Biology, researchers showed that synthetic long-read sequencing using Element Biosciences’ LoopSeq™ technology identified changes in isoform expression that enhanced researchers’ ability to distinguish among cancerous, metastatic, and normal tissues in a study of colon cancer samples. Further inquiry into isoform-specific changes in gene expression may reveal new biomarkers, drug targets, and insights into cancer progression.
LoopSeq provides highly accurate, long-read information on short-read sequencers without needing a dedicated instrument. When used for RNA sequencing, LoopSeq provides full-length isoform data that short reads cannot. This information is particularly valuable for understanding changes in gene expression in cancer, where structural variants, mutation phasing, gene fusions, and aberrant splicing are known to impact disease progression, severity, or treatment response. Better information about this molecular “dark matter” can reveal novel therapeutic targets or serve as biomarkers to improve early detection.
LoopSeq: A Validated Alternative to Native Long Reads
The Nature Communications Biology study was led by Jianhua Luo, MD, PhD, Professor of Pathology at the University of Pittsburgh, and Tuval Ben-Yehezkel, Senior Director of Applications at Element Biosciences.
Luo, Ben-Yehezkel, and colleagues first validated the LoopSeq method by sequencing Hela total RNA spiked with the External RNA Controls Consortium (ERCC) sample and comparing the data to previously published results from long-read sequencers. LoopSeq produced a .01% per base error rate, which was lower than available results from PacBio, Oxford Nanopore, and Illumina sequencers.
Isoforms as Markers of Cancer Progression
The researchers then used LoopSeq to study human colon cancers, sequencing control tissue, primary tumors, and lymph node metastases. The researchers used probe-capture oligos to capture the split regions of the 2,193 most common cancer-related gene fusions found in the TCGA database. With LoopSeq data, the researchers could quantitate differentially expressed genes (DEGs) and differentially expressed isoforms (DEIs) across the three sample types. Remarkably, they found that while hierarchical clustering based on DEGs did not adequately distinguish between tumor and metastatic samples, clusters that instead relied on DEIs did. (Figure 1)
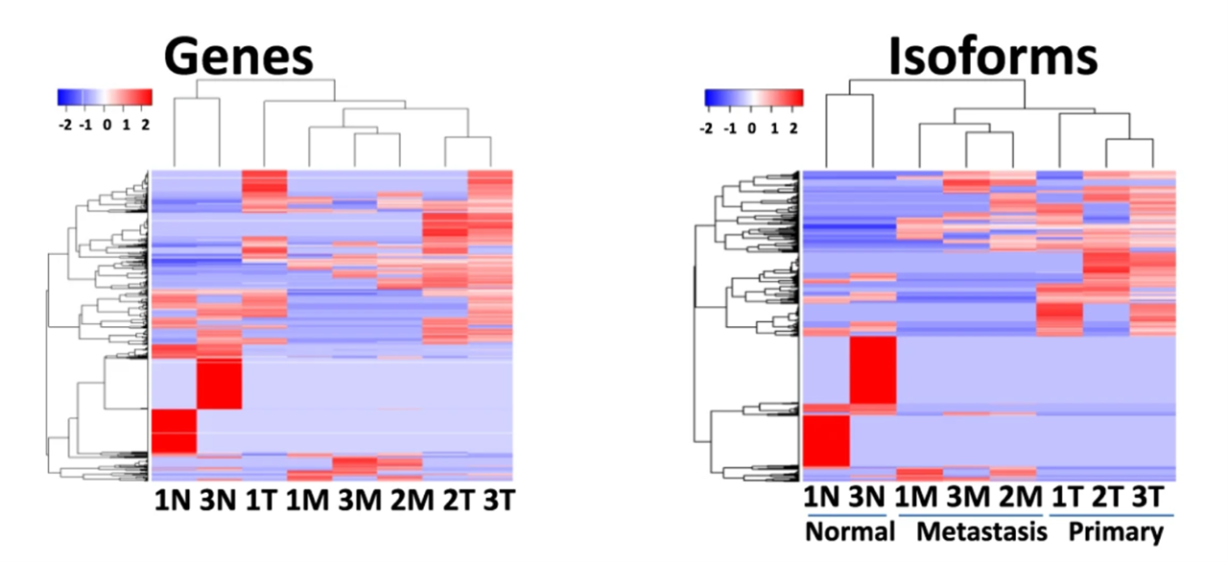

Drilling further into what aspect of gene expression data provides the most information on cancer stage, the authors considered DEGs with unchanged isoform patterns, DEGs with changes in isoform distribution, and DEIs with no net change in gene expression level.
Interestingly, focusing on isoform redistribution without gene expression changes produced the best tissue-differentiation results (Figure 2).
The authors note that “DEIs, which might have previously been inaccessible and were hidden within comparable gene expression levels, represent an additional dimension in differential expression analysis.”
Another notable finding was the detection of single nucleotide variations (SNVs) that were unique to specific isoforms. The researchers found 4,042 SNVs in the six cancer samples. Of the 1,509 SNVs found with at least two isoforms and five assembled contigs, 86% were not distributed evenly.
Finally, four previously unknown fusion genes were found in the LoopSeq data using SQANTI, a bioinformatics pipeline for classifying long reads by splice junctions. One novel fusion, STAMBPL1-FAS, was initially discovered in just two of the metastasis data sets but was later found in all of the study cancer samples using qPCR.
STAMBPL1 is a deubiquitinase inhibitor of apoptosis in the nNF-kB signaling pathway, whereas FAS is a cell surface death receptor.
Isoform Context Expands Our View of Cancer Biology
While quantitative RNA sequencing has long been used to understand the impact of genetic mutations on tumor biology, this proof-of-concept study demonstrates how layering in isoform information can add richness to these analyses. Isoform data can reveal changes in gene expression that are invisible to short-read methods but correlate strongly with the tumor stage. In addition, long-read sequencing continues to reveal new gene fusions, some of which may have wide distribution. LoopSeq provides a new, more accessible option for generating long-read data that can further our understanding of cancer biology.
Contact us to purchase our LoopSeq 16S or Amplicon Sequencing Kits or get a quote for LoopSeq service.
Source for figures: https://www.nature.com/articles/s42003-021-02024-1
Figures 1 and 2 for the blog are Figure 2A and Figure 2C from the paper, respectively.