Sign up for our newsletter
Join our scientific community to stay up to date with Element news, insights, and product updates.
This site is protected by reCAPTCHA and the Google Privacy Policy and Terms of Service apply.
A new approach to measuring sensitivity and specificity across whole genome sequencing data, highlighting the role of AVITI™ sequencing for low error confirmation
Key highlights
-
A new SMaHT benchmark enables accurate measurement of sequencing performance below 1% VAF.
-
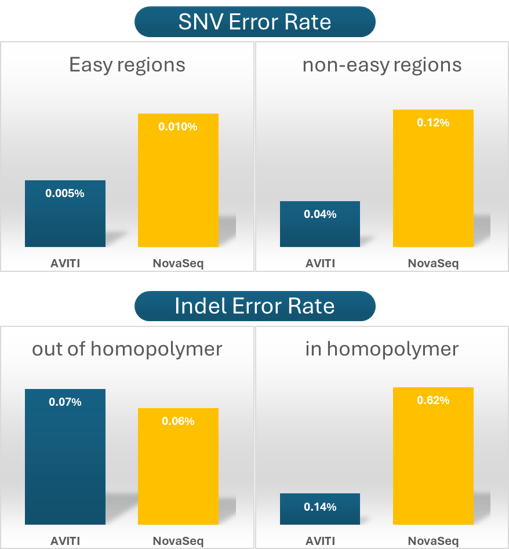
AVITI delivers higher SNV accuracy across both easy and hard genomic regions.
-
Avidite Base Chemistry™ shows ~5× lower indel error than SBS in homopolymer regions.
Measuring true, genome-wide sequencing accuracy is harder than it might seem, especially when the goal is to evaluate sensitivity and specificity at extremely low variant allele frequencies (VAFs). Despite what the label on a control tube may suggest, there is no such thing as a perfectly homogeneous genomic DNA (gDNA) control. Cell lines can accumulate culture-induced mutations, and while synthetic controls created by mixing well-characterized gDNA samples can approximate low-frequency variants, they also introduce distinct mutational haplotypes that may skew results. On top of that, uncharacterized somatic variants can make it difficult to accurately assess specificity.
The Somatic Mosaicism across Human Tissues (SMaHT) network was created to address these challenges, with the goal of cataloging somatic mutations across 10–15 tissues from 150 donors. In a recent preprint, Accurate detection of sub 1% frequency somatic mutations by whole genome sequencing, Jing et al. describes a new approach developed within the SMaHT framework that overcomes many of the limitations of traditional control strategies. Their work introduces a practical and reliable way to measure sequencing accuracy at variant allele frequencies below 1%.
Why sub 1% somatic mutation detection is so difficult
Somatic mosaicism is common across development, aging, and disease, but most somatic mutations are rare within any given tissue. A mutation present at 0.5% VAF may appear in only 1 out of 200 reads. At that level, even small sequencing or alignment errors can overwhelm the true biological signal.
The challenge is compounded by genomic context. Regions with repeats, segmental duplications, or low sequence complexity, often referred to as “non easy” regions, are more prone to mapping ambiguity and background noise. As a result, detection can vary dramatically depending on where a variant occurs in the genome.
A biological benchmark built for confidence
Somatic mutations naturally accumulate over time due to environmental exposure and aging, and skin tissue is known to carry a relatively high mutational burden. A cross-institutional research team took advantage of this biology to generate both positive and negative control variants from a single individual. Starting with a skin biopsy, individual fibroblasts were isolated and reprogrammed into induced pluripotent stem cell (iPSC) lines. These were then expanded into clonal sublines and sequenced, allowing true somatic mutations in each clone to be identified with high confidence.
To create a set of negative controls, a second biopsy was taken from a different area of the same individual’s skin. Subclones from this biopsy were isolated and sequenced in the same way. Because somatic mutations arise independently in different skin regions, variants found in one biopsy would be statistically unlikely to appear in the other. Together, this strategy produced a well-defined set of positive and negative control variants spanning a range of allele frequencies, exactly what’s needed to evaluate sequencing accuracy across platforms.
Illumina sequencing-by-synthesis (SBS) has long been the standard for generating the deep whole-genome data required to detect low-frequency somatic variants. In this study, high-coverage NovaSeq data were compared with data generated using newer sequencing technologies, including the AVITI platform. While Element’s Avidite Base Chemistry (ABC™) has already been shown to deliver higher per-base accuracy than SBS in several studies, its performance at VAFs below 1% had not been widely explored.
AVITI shows low error where it matters most
The results continue to support a growing body of evidence showing that our technology can exceed accuracy levels that have remained largely unchanged in short-read sequencing for more than a decade. Single nucleotide variants (SNVs) were analyzed in both “easy” and “non-easy” regions of the genome, as defined by 1000 Genomes Project masks that highlight challenging, repeat-rich areas. Indel error rates were also evaluated in both homopolymer and non-homopolymer contexts.
Two results stand out:
-
Across these analyses, AVITI showed consistently higher SNV accuracy regardless of genomic region.
-
Indels are particularly difficult to call at low VAF because errors are enriched in and around homopolymers. In this study, AVITI showed substantially lower indel mismatch rates than other platforms, including approximately fivefold lower error than NovaSeq in homopolymer regions.
Why this matters
Measuring true, genome-wide sequencing accuracy is harder than it might seem, especially when the goal is to evaluate sensitivity and specificity at extremely low VAFs. Despite what the label on a control tube may suggest, there is no such thing as a perfectly homogeneous genomic DNA (gDNA) control. Cell lines can accumulate culture-induced mutations, and while synthetic controls created by mixing well-characterized gDNA samples can approximate low-frequency variants, they also introduce distinct mutational haplotypes that may skew results. On top of that, uncharacterized somatic variants can make it difficult to accurately assess specificity.
As evidence continues to build around the advantages of ABC chemistry over legacy sequencing approaches, adoption of the AVITI platform is expanding in applications that demand both high sensitivity and specificity. From tumor profiling to forensics, moving beyond traditional SBS sequencing is helping push the boundaries of what’s detectable in complex samples.
See how other researchers are using AVITI sequencing in their publications.